IgA nephropathy (IgAN) is the most common form of primary glomerulonephritis worldwide and the leading cause of end-stage kidney failure in East Asia.
In IgA nephropathy (IgAN) is diagnosed most commonly in the second or third decade of life. The kidney biopsy shows predominant deposition of IgA-containing immune complexes in the glomerular mesangium. These immune complex deposition leads to glomerulonephritis, glomerular sclerosis, and progressive loss of kidney function.
The etiology of IgAN is poorly understood and the genetic architecture is complex. Based on the current pathogenesis model, the disease occurs in the setting of inherited defect that results in exaggerated production of IgA1 with aberrant O-glycosylation of its hinge region. This aberrantly glycosylated IgA1 is targeted by an IgA or IgG autoimmune response, resulting in production of immune complexes that injure the kidney. We have shown that the O-glycosylation defect is heritable and genome-wide association studies (GWAS) have implicated common variants in C1GALT1 and C1GALT1C1 in levels of aberrantly glycosylated IgA1. In addition, we have performed many GWAS’s in individuals of European and Asian ancestry and identified 15 significant associations for IgAN in HLA, ITGAM-ITGAX, VAV3, CARD9, CFHR1, DEFA, TNFSF13 loci.These studies have implicated the critical role of the intestinal immune system for IgA production, the alternative complement pathway and APRIL/TACI in IgAN pathogenesis. These three pathways have now been successfully targeted for therapy and are expected to transform care for IgAN patients. These same studies nominate novel therapeutic targets that may be efficacious for subgroup of patients. We anticipate that our future studies will continue to reclassify IgAN and unravel disease pathogenesis, and simultaneously identify individuals at the highest risk of progression, and enable more precise diagnosis and treatment.
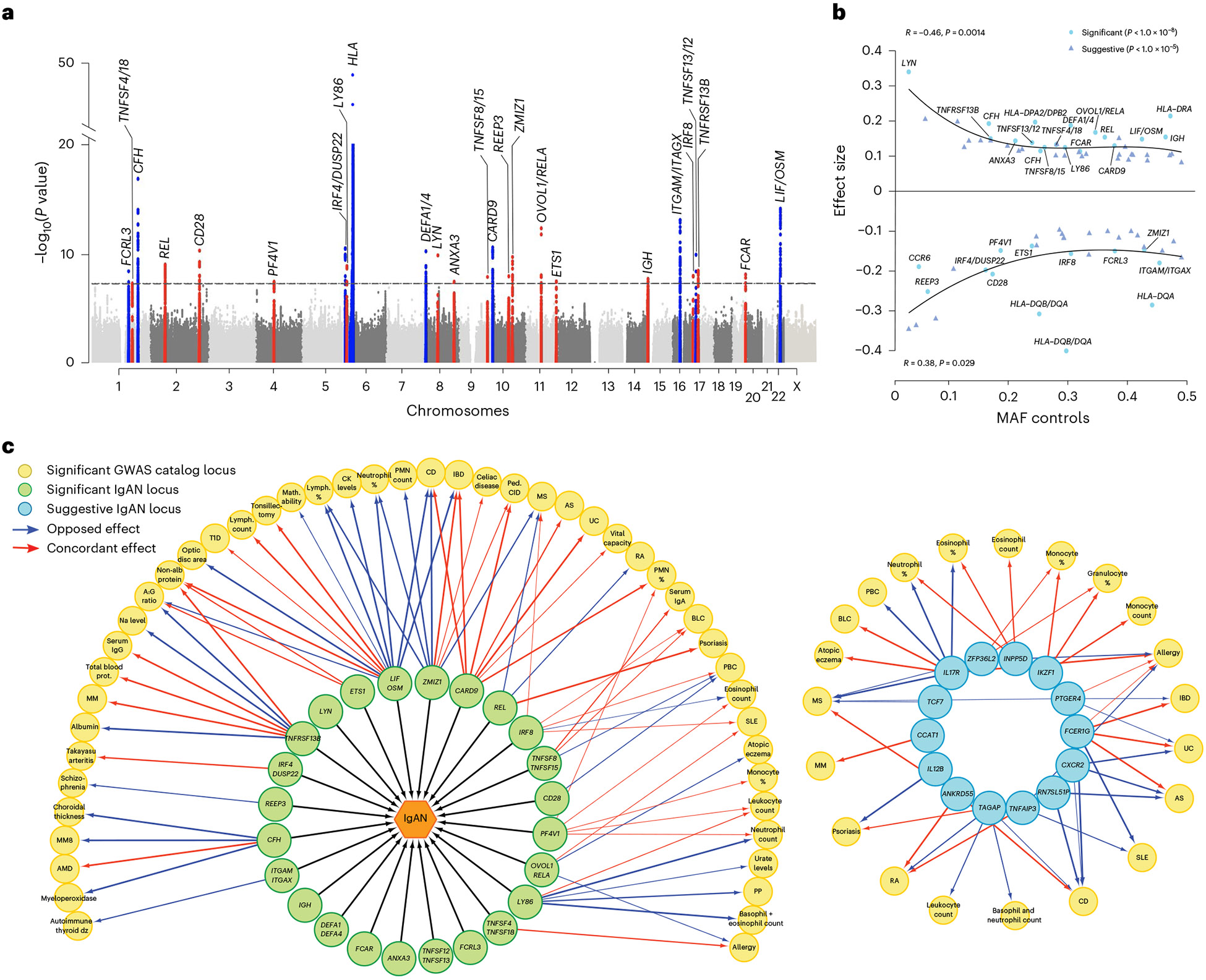
We are now engaged in larger GWAS and genome and exome sequencing projects in IgAN. These studies, conducted in >30,000 individuals, are identifying new loci for IgAN. In parallel, we are studying immunological mechanisms leading to activation of IgA producing cells and resultant increased production of galactose deficient IgA1. These studies are highlighting new pathways such as loci encoding cytokines and their cognate receptors, and dysregulated immune cell types that may be promising targets for therapy. Our latest functional studies demonstrate that a abnormalities in O-glycosylation alter mucosal immunity and B lymphocyte homing, pointing to an expanded role of aberrant O-glycosylation in the pathogenesis of IgAN.